By Hunter Martinez
The flow cytometer is a critical piece of equipment in any immunologist’s arsenal. Flow cytometry allows for the analysis of 4 to 6x105 cells per minute, each resolved at the single cell level. Scientists can interrogate protein, gene expression, as well as metabolic state of a cell using a flow cytometer.
The Basics of Cytometry
A cytometer accomplishes this feat using the principle of fluorescence. Fluorescence occurs when light from a laser is absorbed by a fluorophore, and then emitted as longer wavelength light or lower energy photon. In a flow experiment, fluorophores are conjugated to antibodies and mixed with cells of interest. Multiple antigens can be detected simultaneously, with each antigen assigned a specific fluorophore. However, fluorophores do not emit light in discrete peaks at one wavelength. Rather, the emission peaks are usually broad, spanning several wavelengths along the visible light spectrum. In the early days of cytometry, when only one or two fluorochromes were used to analyze cells, the emission spectrum of the fluorophore was not an obstacle for analysis, as overlap was rare. Increasingly, scientists are seeking to analyse more parameters within a single sample and overlapping emission spectra present a problem: how do you discern which emission came from which fluorophore in a multiparameter experiment? There are two major approaches available to tackle this issue: conventional and spectral cytometry.
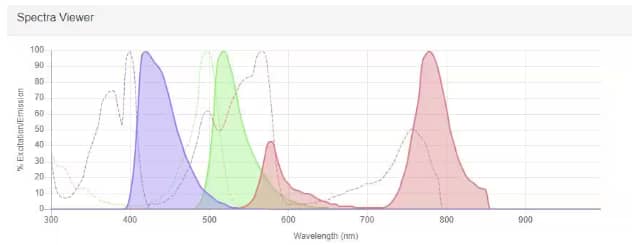
Spectra Viewer example readout of 3 different fluorophores: Alexa Fluor 405 (purple), Alexa Fluor 488 (green), and PE-Cy7 (red). The excitation spectrum for each is displayed as a dotted line whereas the emission spectrum is shown as a solid line with the area filled in under the curve.
A Narrow View: Conventional Cytometry
Conventional flow cytometers approach the fluorescence emission “problem” by filtering the emission light through bandpass filters allowing specific wavelengths of light through to each detector and then “subtracting” light spillover from other fluorophores using compensation. Compensation, or subtracting the spillover light, is accomplished by recording the two overlapping parameters separately and mathematically deducting the overlapping areas on the spectrum upon their mixing. Filters typically correspond to the peak emission wavelength of fluorophores used in each experiment. This means a conventional cytometer can only obtain data from as many fluorescent parameters as there are detectors. Current panel limit for a conventional cytometer is 15-20 colors.
The Whole Picture: Spectral Cytometry
A spectral cytometer takes a more holistic approach, and importantly, removes this notion of a “primary” channel for each fluorophore. A spectral cytometer is set up with many detectors to monitor small segments across the full light spectrum. Collectively, each detector is assumed to be detecting a mixture of photons from multiple fluorophores in any given experiment. Rather than compensating or subtracting the spillover to correct fluorophore overlap, the mathematics of mixture modeling, specifically principal component analysis and least squares unmixing, are used to resolve each parameter. Like the conventional cytometer, the spectral cytometer must see each fluorescent parameter by itself in addition to the mixed unknown sample. One additional benefit of spectral cytometry is the subtraction of cellular autofluorescence to improve signal resolution. Flow panels on spectral cytometers can have as many as 40 colors, but panels must be carefully designed.
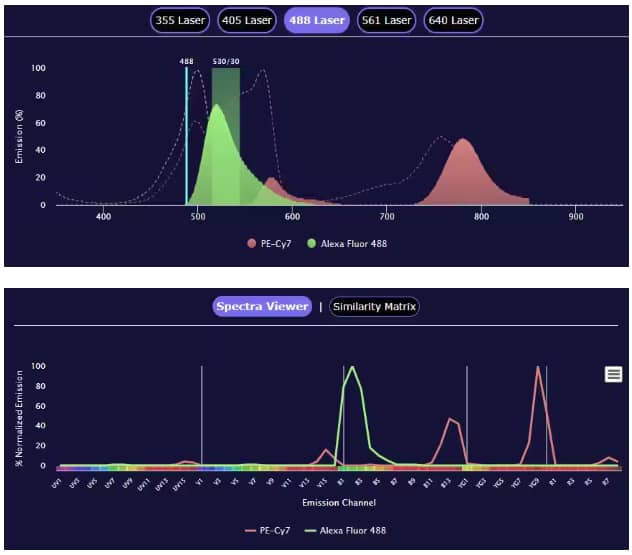
Flow cytometry panel builder tool comparing the spectra view readout of conventional flow cytometry (top) and spectral flow cytometry (bottom). The example conventional flow panel was built using the BD LSR2 Fortessa X-20 (5 Laser), shown under the 488 laser, while the spectral flow panel used the Cytek Aurora 5L instrument. Both examples use PE-Cy7 conjugated to Mouse Anti-Human CD3 epsilon Monoclonal Antibody (Catalog # NBP1-30426PECY7) and Alexa Fluor 488 conjugated to Mouse Anti-Human CD4 Monoclonal Antibody (Catalog # FAB39713G).
Shared Characteristics
Both approaches in cytometry still require intelligent panel design. When considering fluorophores that are bright (e.g. APC, PE), it is recommended to assign them to lowly expressed antigens. Additionally, avoid using fluorophores with similar spectra (or peak emissions) for antigens expressed on the same cell. Finally, both modern versions of these cytometers have automated high-throughput capabilities in the form of plate readers.
Bio-Techne Resources for Flow Cytometry:
- Flow Cytometry Panel Builder Tool
- Spectra Viewer
- Flow Cytometry Handbook
- Cell Marker Tool (at R&D Systems)
- Cell Marker Guide Poster
- Multicolor Flow Cytometry Kits and Panels

Hunter Martinez, PhD
Stanford University School of Medicine
-
Novo, D. (2022) A comparison of spectral unmixing to conventional compensation for the calculation of fluorochrome abundances from flow cytometric data Cytometry
-
Szalóki G, Goda K. (2022) Compensation in multicolor flow cytometry Cytometry A. 87:982-985.
-
Nolan, J.P. (2022) The evolution of spectral flow cytometry Cytometry A 101:812-817.
-
Novo D, Grégori G, Rajwa B. (2013) Generalized unmixing model for multispectral flow cytometry utilizing nonsquare compensation matrices Cytometry A. 83:508-520.