By Christina Towers, PhD
Autophagy flux: Basic principles
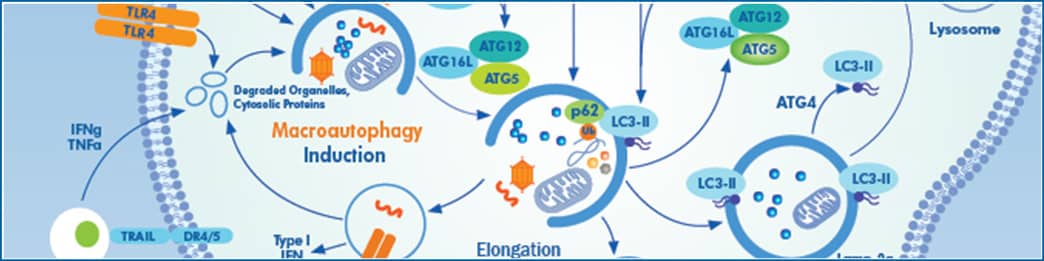
Autophagic flux is defined as the amount of cellular material degraded and recycled through the process of autophagy, whereby cells break down and discard waste. Briefly, the autophagy process involves the formation of an autophagosome, which engulfs cytoplasmic materials. Fusion of the autophagosome with acidic lysosomes mediates the degradation of cellular contents. The process involves over 30 core autophagy proteins (ATG proteins) that mediate different stages in the pathway, including autophagosome induction and formation, while others regulate autophagosomal closure and/or fusion with lysosomes. Flux through the pathway is often measured by pharmacologically inhibiting lysosomal function with drugs like chloroquine or bafilomycin-A and monitoring the accumulation of autophagy-targeted proteins like LC3-II or p62. It is particularly critical to monitor flux, i.e. accumulation of lysosomal inhibitors, versus levels of LC3-II. LC3-II protein expression increases with induction of autophagy because it is a necessary component of functional autophagosomes; yet LC3-II will also accumulate when autophagy is inhibited because it gets degraded as the autophagosome fuses with the acidic lysosome. Steady-state assays should be combined with autophagic flux measuring assays to distinguish between the two aforementioned autophagy mechanisms involving LC3-II.
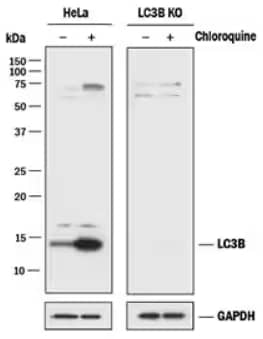
LC3B Knockout Validation: LC3B Antibody [NB100-2220] - Lysates of HeLa parental cell line and LC3B knockout HeLa cell line (KO) untreated (-) or treated (+) with 50 uM Chloroquine for 18 hours. Polyvinylidene difluoride (PVDF) membrane was probed with 0.5 ug/mL of Rabbit Anti-LC3B Polyclonal Antibody [NB100-2220] followed by HRP-conjugated Anti-Rabbit IgG Secondary Antibody [HAF008]. A specific band was detected for LC3B at approximately 15 kDa in the parental HeLa cell line but is not detectable in the knockout HeLa cell line. GAPDH is shown as a loading control. This experiment was conducted under reducing conditions.
Autophagic flux modulation: Targeting LC3-II formation
To genetically interfere with this process, shRNA mediated knock down or CRISPR/Cas9 mediated knock out of core autophagy genes (ATGs) can be used; however, it is important to understand the consequences of targeting the different core ATG proteins. Classically, ATG proteins necessary for LC3-I conjugation to the lipid phosphatidylethanolamine (PE), including ATG5, ATG7, ATG3, or ATG12, have been targeted to functionally inhibit autophagic flux. Efficient loss of any of these proteins will eliminate LC3-II formation; however, autophagosomes will still be formed. These LC3-deficient structures can still fuse with lysosomes but with a drastic decrease in efficiency, resulting in a significant reduction in autophagic flux. Measuring how much flux is affected can be difficult, since LC3-II levels +/- lysosomal inhibitors can no longer be used in this context. Instead, one should consider using p62 accumulation or the recently created STX17-GFP construct, which includes an autophagic-lysosomal SNARE protein that marks autophagosomes without affecting their functionality and green fluorescent protein. Notably, many of these proteins have autophagy-independent functions; for example, ATG5 and ATG7 are also necessary for LC3 associated phagocytosis (LAP), and FIP200 plays an important role in TNFα-induced apoptosis.
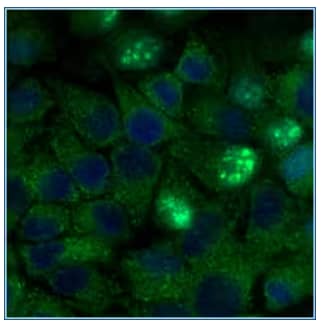
Immunocytochemistry/Immunofluorescence: ATG5 Antibody [NB110-53818] - HeLa cells were fixed for 10 minutes using 10% formalin and then permeabilized for 5 minutes using 1X PBS + 0.05% Triton-X100. The cells were incubated with anti-ATG5 at 2 ug/ml overnight at 4C and detected with an anti-rabbit Dylight 488 [NB110-53818G] (Green) at a 1:500 dilution. Nuclei were counterstained with DAPI [NBP2-31156-1mg] (Blue). Cells were imaged using a 40X objective.
Autophagic flux modulation: Targeting autophagosome formation
Alternatively, to completely inhibit autophagosome formation, the pathway can be targeted further upstream at the stage of phagophore initiation and nucleation. This includes proteins that comprise the FIP200-RB1CC1 complex. For example, loss of the FIP200-ATG13 interaction has been shown to abolish the formation of autophagosomes completely. In this context, LC3-II conjugation remains intact as a readout for autophagy inhibition, and a complete block of flux is expected. As mentioned above, many of the proteins involved in this process have autophagy-independent functions, which are also lost when these upstream genes are targeted.
Wherever one chooses to genetically interfere with the autophagic pathway, understanding the correct assays needed to confirm inhibition and being aware of the off-target, i.e. autophagy-independent functions, of each targeted gene is essential.

Christina Towers, PhD
University of Colorado (AMC)
Dr. Towers studies the roles of autophagy, apoptosis and cell death in cancer.
-
Chen, S. et al. (2016) Distinct roles of autophagy-dependent and -independent functions of FIP200 revealed by generation and analysis of a mutant knock-in mouse model Genes Dev 30:856-869.
-
Klionsky, D.J. et al. (2016) Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition) Autophagy 12:1-22.
-
Martinez, J. et al. (2011) Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells Proc Natl Acad Sci U S A 108:17396-17401.
-
Tsuboyama, K. et al. (2016) The ATG conjugation systems are important for degradation of the inner autophagosomal membrane Science 354:1036-1041.